Синдром Альпорта (семейный гломерулонефрит)
– это редкое генетическое заболевание, которое характеризуется гломерулонефритом, прогрессирующей почечной недостаточностью, нейросенсорной тугоухостью и поражением глаз.
Заболевание было впервые описано британским врачом Артуром Альпортом в 1927 году.
Синдром Альпорта встречается очень редко, но в США он отвечает за 3% случаев терминальной почечной недостаточности у детей и 0,2% у взрослых, а также считается наиболее распространенным типом семейного нефрита.
Тип наследования синдрома Альпорта может быть разным:
• Х-сцепленный доминантный (XLAS): 85%. • Аутосомно-рецессивный (ARAS): 15%. • Аутосомно-доминантный (ADAS): 1%.
Наиболее распространенная Х-сцепленная форма синдрома Альпорта приводит к терминальной стадии почечной недостаточности у мужчин. Гематурия обычно возникает у мальчиков с синдромом Альпорта в первые годы жизни. Протеинурия обычно отсутствует в детстве, но это состояние часто развивается у мужчин с XLAS и у представителей обоих полов с ARAS. Потеря слуха и поражение глаз никогда не обнаруживаются при рождении – они возникают в позднем детстве или в юности, незадолго до развития почечной недостаточности.
Причины и механизм развития синдрома Альпорта
Синдром Альпорта вызван мутациями в генах COL4A4, COL4A3, COL4A5, отвечающих за биосинтез коллагена. Мутации в указанных генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах.
Базальные мембраны – это тонкие пленочные структуры, которые поддерживают ткани и отделяют их друг от друга. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта.
Симптомы заболевания
Наследственный нефрит возникает при нехватке в организме коллагена — важного элемента в структуре соединительной ткани. Вследствие коллагенового дефицита происходит истончение и постепенное разрушение базальных мембран в почечных клубочках, внутреннем ухе и глазном аппарате. Органы перестают полноценно функционировать. В почечных клубочках нарушается процесс фильтрации, из крови в мочу попадают белок и эритроциты. Заболевание носит прогрессирующий характер с нарастанием симптомов почечной недостаточности, что может вызвать летальный исход.
Впервые заболевание проявляется в детском возрасте. Начало недуга может иметь скрытые признаки. О наличии гематурии часто становится известно случайно на фоне респираторных вирусных инфекций, протекающих с тонзиллитом или фарингитом. Период между началом ОРВИ и появлением гематурии составляет несколько дней (обычно 1–2). По этому признаку можно отличить данное заболевание от инфекционного гломерулонефрита.
К внепочечным симптомам относятся:
- тугоухость, обусловленная невритом слухового нерва;
- ухудшение зрения, связанное с катарактой, изменением формы хрусталика, появлением белых или желтых вкраплений на сетчатке в районе макулы, миопией, кератоконусом;
- задержка в психофизическом развитии;
- врожденные дефекты — высокое небо, синдактилия, эпикант, деформация ушей, патологии прикуса;
- лейомиоматоз пищевода, трахеи, бронхов.
К неспецифическим общеинтоксикационным признакам патологии относятся:
- головная боль,
- миалгия,
- головокружение,
- резкие колебания артериального давления,
- одышка,
- частое, поверхностное дыхание,
- шум в ушах,
- бледность кожи,
- частые позывы к мочеиспусканию,
- диспепсия,
- ухудшение аппетита,
- нарушение режима сна и бодрствования,
- зуд кожи,
- судороги,
- боль в груди,
- спутанность сознания.
Клиника
Гематурия – это наиболее частое и раннее проявление синдрома Альпорта. Микроскопическая гематурия наблюдается у 95% женщин и практически у всех мужчин. У мальчиков гематурию обычно выявляют в первые годы жизни. Если у мальчика за первые 10 лет жизни не обнаружена гематурия, то американские эксперты рекомендуют считать, что у него маловероятно наличие синдрома Альпорта.
Протеинурия в детстве обычно отсутствует, но иногда развивается у мальчиков с Х-сцепленным синдромом Альпорта. Протеинурия, как правило, прогрессирует. Значительная протеинурия у больных женского пола встречается нечасто.
Гипертензия чаще присутствует у пациентов мужского пола с XLAS и у больных обоих полов с ARAS. Частота и тяжесть гипертензии повышается с возрастом и по мере прогрессирования почечной недостаточности.
Нейросенсорная тугоухость (нарушение слуха) – это характерное проявление синдрома Альпорта, которое наблюдается довольно часто, но не всегда. Есть целые семьи с синдромом Альпорта, которые страдают от тяжелой нефропатии, но имеют нормальный слух. Нарушение слуха никогда не обнаруживается при рождении. Билатеральная высокочастотная нейросенсорная тугоухость обычно проявляется в первые годы жизни или в раннем подростковом возрасте. На ранней стадии болезни нарушение слуха определяется только при аудиометрии.
По мере прогрессирования, нарушение слуха распространяется на низкие частоты, включая человеческую речь. После появления тугоухости следует ожидать вовлечения почек. Американские ученые утверждают, что при Х-связанном синдроме Альпорта 50% мужчин страдают нейросенсорной тугоухостью к 25 годам, а к 40 годам – около 90%.
Передний лентиконус (выпячивание центрального участка хрусталика глаза вперед) наблюдается у 25% пациентов с XLAS. Лентиконуса нет при рождении, но с годами он приводит к прогрессирующему ухудшению зрения, которое заставляет больных часто менять очки. Состояние не сопровождается болью в глазах, покраснением или нарушениями цветового зрения.
Ретинопатия – это самое распространенное проявление синдрома Альпорта со стороны органа зрения, поражает 85% мужчин с Х-сцепленной формой болезни. Появление ретинопатии обычно предшествует почечной недостаточности.
Задняя полиморфная дистрофия роговицы – редкое состояние при синдроме Альпорта. У большинства нет никаких жалоб. Мутация L1649R в гене коллагена COL4A5 может также вызывать истончение сетчатки, которое ассоциируется с Х-сцепленным синдромом Альпорта.
Диффузный лейомиоматоз пищевода и бронхиального дерева – еще одно редкое состояние, которое наблюдается в некоторых семьях с синдромом Альпорта. Симптомы появляются в позднем детском возрасте и включают нарушение глотания (дисфагия), рвоту, боль в эпигастрии и за грудиной, частые бронхиты, одышку, кашель. Лейомиоматоз подтверждается компьютерной томографией или МРТ.
Клиническая картина
Синдром Альпорта протекает с почечными и внепочечными признаками. Симптомы Альпорта у детей чаще всего развиваются в возрасте от 5 до 10 лет. В этот период появляется кровь и белок в моче. У мальчиков гематурия может быть диагностирована в течение первого года жизни. При отсутствии клинической картины до 10 лет принято считать, что вероятность развития патологии стремится к нулю.
При развитии протеинурии возрастает риск развития нефротического синдрома, для которого характерны:
- повышенная обрюзглость тела;
- рост артериального давления;
- общая слабость и быстрая утомляемость;
- жажда;
- снижение аппетита;
- болевой синдром в области почек;
- нарушения со стороны органов пищеварения (тошнота, рвота, диарея, метеоризм);
- снижение суточного объема урины;
- бледность и сухость кожных покровов;
- одышка.
Диагностика синдрома Альпорта
• Лабораторные анализы. Анализ мочи: у больных с синдромом Альпорта чаще всего присутствует кровь в моче (гематурия), а также высокое содержание белка (протеинурия). Анализы крови демонстрирует почечную недостаточность. • Биопсия тканей. Ткань почек, полученную при биопсии, исследуют с помощью электронной микроскопии на наличие ультраструктурных аномалий. Биопсия кожи менее инвазивна, и американские эксперты рекомендуют выполнять ее в первую очередь. • Генетический анализ. В диагностике синдрома Альпорта, если остаются сомнения после биопсии почки, генетический анализ используется для получения однозначного ответа. Определяются мутации генов синтеза коллагена типа IV. • Аудиометрия. Все дети с семейной историей, позволяющей заподозрить синдром Альпорта, должны проходить высокочастотную аудиометрию для подтверждения нейросенсорной тугоухости. Рекомендуется периодический мониторинг. • Обследование глаз. Обследование у офтальмолога очень важно для раннего выявления и мониторинга переднего лентиконуса и других аномалий. • УЗИ почек. На поздних стадиях синдрома Альпорта ультразвуковое исследование почек помогает выявить структурные нарушения.
Британские специалисты, основываясь на новых данных (2011) по генетическим мутациям у пациентов с Х-сцепленным синдромом Альпорта, рекомендуют анализ на мутации гена COL4A5, если пациент отвечает хотя бы двум диагностическим критериям по Gregory, и анализ COL4A3 и COL4A4, если мутация COL4A5 не обнаружена или подозревается аутосомное наследование.
Прогноз и профилактика
Прогноз наследственного нефрита всегда серьезен.
Прогностически неблагоприятными критериями течения наследственного нефрита являются:
- мужской пол;
- раннее развитие хронической почечной недостаточности у членов семьи;
- протеинурия (более 1 г/сут);
- утолщение гломерулярных базальных мембран по данным микроскопии;
- неврит слухового нерва;
- делеция в гене Соl4А5.
Прогноз доброкачественной семейной гематурии более благоприятен.
Лечение синдрома Альпорта
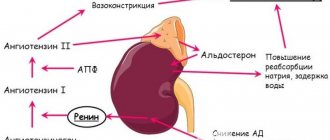
Синдром Альпорта пока неизлечим. Исследования показали, что ингибиторы АПФ могут уменьшать протеинурию и замедлять прогрессирование почечной недостаточности. Таким образом, использование ИАПФ целесообразно у пациентов с протеинурией, независимо от наличия гипертензии. То же самое касается антагонистов АТII-рецепторов. Оба класса препаратов, судя по всему, помогают уменьшить протеинурию путем снижения внутриклубочкового давления. Более того, ингибирование ангиотензина-II, ростового фактора, отвечающего за гломерулярный склероз, теоретически может замедлять склерозирование.
Некоторые исследователи предполагают, что циклоспорин способен уменьшать протеинурию и стабилизировать почечную функцию у пациентов с синдромом Альпорта (исследования были небольшими). Но отчеты говорят, что ответ пациентов на циклоспорин очень вариабельный, и иногда препарат может ускорять интерстициальный фиброз.
При почечной недостаточности стандартная терапия включает эритропоэтин для лечения хронической анемии, препараты для контроля остеодистрофии, коррекцию ацидоза и антигипертензивную терапию для контроля АД. Применяется гемодиализ и перитонеальный диализ. Больным с синдромом Альпорта трансплантация почки не противопоказана: опыт пересадки в США показал хорошие результаты.
Генная терапия при разных формах синдрома Альпорта является перспективным вариантом лечения, который сегодня активно изучается западными медицинскими лабораториями.
Константин Моканов: магистр фармации и профессиональный медицинский переводчик
Этиология
Этиологию наследственного нефрита до сих пор невозможно полностью установить. Наиболее вероятной причиной принято считать мутацию гена, который отвечает за синтез белков в почечной ткани.
Развитию патологического процесса могут поспособствовать следующие факторы:
- тяжёлые инфекционные заболевания;
- чрезмерные физические нагрузки;
- прививки.
В медицинской практике встречались случаи, когда развитие наследственного нефрита могло спровоцировать даже обычное ОРВИ. Поэтому дети, которые имеют генетическую предрасположенность, должны проходить полное обследование чаще.
Примечательно то, что наследственный нефрит имеет доминантный тип наследования. Это значит, что если носителем будет мужчина, то у него родится здоровым только сын. Дочь будет не только носителем гена, но и будет его передавать как сыновьям, так и дочерям.
3. Лечение гематурии
Возможно наблюдение при контроле почечной функции, поскольку в большом проценте случаев гематурия может спонтанно исчезнуть.
Терапевтическая тактика зависит от причин гематурии.
3.1 Консервативное лечение
IgA-нефропатия:
- без протеинурии — контроль уровня протеинурии и почечных функций в АПУ;
- при значительной протеинурии — преднизолон 1-2 мг/кг или цитостатики 14-21 дня в стационаре, далее под контролем анализов в АПУ;
- полиненасыщенные жирные кислоты (Омега-3 триглицериды);
- антикоагулянты и антиагреганты;
- длительно иАПФ фозиноприл, эналаприл в среднем 0,1-0,3 мг/кг.
Синдром Альпорта:
- иАПФ фозиноприл, эналаприл с индивидуальным подбором дозы, в среднем 0,1-0,3 мг/кг по фозиноприлу.
Болезнь тонких базальных мембран:
- благоприятное течение;
- не рекомендовано проведение терапии.
Постинфекционный гломерулонефрит:
- антибиотик пенициллинового ряда 14 дней;
- при использовании антибиотиков в предшествующие 1-3 мес.– защищенные аминопенициллины: амоксициллин+клавулановая кислота;
- диуретики: чаще фуросемид, реже спиронолактон;
- иАПФ фозиноприл, эналаприл с индивидуальным подбором дозы;
- блокаторы медленных кальциевых каналов (GPPs): амлодипин или лаципидил с подбором дозы;
- прогноз в 90% случаев благоприятный;
- редкие варианты с экстракапиллярными изменениями и ХПН могут потребовать диализа, пульс-терапии метилпреднизолоном;
- среднее пребывание в стационаре 14-21 день (при отсутствии осложнений).
Нефрит Шенлейн-Геноха:
- в активной стадии преднизолон 1-1,5 мг/кг с вариабельной длительностью;
- при изолированной хронической гематурии не рекомендовано назначение терапии;
- при экстракапиллярных изменениях (полулуния) пульс-терапия метилпреднизолоном 30мг/кг с последующим в/в циклофосфамидом 15-20 мг/кг 1 раз в месяц полгода;
- длительность госпитализации определяется тяжестью течения болезни;
- повторные введения циклофосфамида можно проводить в стационаре одного дня;
- при ремиссии прогноз благоприятный.
Идиопатическая гиперкальциурия:
- не целесообразна диета со сниженным содержанием кальция;
- увеличить объем принимаемой жидкости;
- при упорном течении и риске образования конкрементов целесообразен прием 1мг/кг/день гидрохлоротиазида и цитрата блемарена под контролем рН мочи 6,2-6,8;
- тиазидные диуретики способствуют электролитным нарушениям вследствие реабсорбции кальция.
Мочекаменная болезнь:
- без спонтанного отхождения конкрементов более 5 мм рекомендована литотрипсия;
- необходимо добиться полного отхождения камней;
- исследование состава камня при рентгеновской дифракции или спектрофотометрии;
- состав конкремента определяет диагностику, метафилактику и диетотерапию;
- при оксалатно-кальциевых и камнях из мочевой кислоты назначение цитратов на фоне обильного приема жидкости.
3.2 Хирургическое лечение
Не требуется
Причины возникновения болезни
Синдром Альпорта — это следствие генетической мутации. В результате её развития в организме начинает синтезироваться аномальный коллаген, который входит в состав почечных мембран и канальцев, органов слуха и зрения, что обуславливает появление клинической симптоматики. Существуют 3 типа наследования заболевания:
- аутосомно-доминантный (если страдает хотя бы один из родителей, недуг 100% будет у ребёнка);
- аутосомно-рецессивный (для рождения больного малыша необходим союз двух людей, являющихся носителями мутантного гена);
- сцепленный с половой хромосомой (передаётся от матери плоду преимущественного мужского пола, так как девушки имеют генотип XX, а мужчины — XY).
Какие факторы, воздействующие на беременную женщину, увеличивают вероятность развития недуга:
- облучение (рентгеновское, ультрафиолетовое);
- радиация;
- проживание в экологически неблагоприятных условиях (загрязнение воды, почвы, атмосферы);
- самовольный приём фармацевтических препаратов (в особенности антибиотиков, гормонов, цитостатиков, иммунодепрессантов);
- работа на вредном производстве (контакт с ядовитыми газами, тяжёлыми металлами);
- употребление наркотиков;
- алкоголизм и курение.
Как лечится болезнь
Специфических препаратов, направленных на лечение данного недуга, не существует. Лекарственные препараты следует сочетать со специальным диетическим питанием. Лечение направлено на нормализацию функционирования почек. На начальных стадиях не требуется медикаментозной терапии. Для детей предусмотрены следующие терапевтические меры:
- освобождение ребенка от физических нагрузок, в т. ч. от уроков физической культуры;
- регулярные прогулки на свежем воздухе;
- строгое соблюдение назначенной врачом диеты (обычно ее необходимо соблюдать в течение всей жизни);
- для устранения симптомов гематурии назначается фитотерапия: настои крапивы, тысячелистника, черноплодной рябины;
- курсовой прием витаминов А, Е, В6, что позволит улучшить общий обмен веществ в организме.
В дальнейшем по мере прогрессирования болезни проводится симптоматическое лечение. Используются такие препараты, как ингибиторы АПФ, блокаторы ангиотензиновых рецепторов, средства для коррекции ацидоза. Для замедления деструктивных процессов и улучшения самочувствия пациентов, рекомендуется прием следующих препаратов:
- Фуросемид (для больных, у которых не нарушен диурез);
- внутривенное введение физраствора (для профилактики обезвоживания);
- Верошпирон, глюкоза и кальция глюконат — для восстановления минерального обмена;
- витамины группы Д;
- анаболические гормоны и железосодержащие препараты, необходимые для ускоренного образования эритроцитов.
При необходимости применяется гемодиализ. В особо тяжелых случаях может потребоваться операция по пересадке почки. Детям подобные вмешательства возможно осуществлять не ранее 15–18 лет. После трансплантации состояние пациентов становится гораздо лучше, особенно если нефрит протекал без нарушений со стороны органов чувств.
Диета при синдроме Альпорта
Лечение недуга надо обязательно проводить в сочетании с диетой. Из рациона следует исключить следующие продукты:
- все копчености и пряности;
- слишком соленую и жирную пищу;
- острые приправы;
- алкогольные напитки;
- продукты с ненатуральными красителями.
Пища должна содержать достаточное количество калорий и витаминов. Белок должен присутствовать, но не в очень больших количествах. Для каждого пациента диета разрабатывается индивидуально с учетом функциональных способностей почек. Наблюдение за пациентом осуществляется в течение всей жизни.