Тубулопатии представляют собой гетерогенную группу болезней мочевыделительной системы, связанных с нарушением процессов реабсорбции, локализующихся в почечных канальцах. Клиническая картина тубулопатии у детей крайне разнообразна. Заболевания проявляются симптомами дефицита определенного микроэлемента: слабостью, мышечной гипотонией, обезвоживанием, полиурией, симптомами рахита, изменениями артериального давления в любую сторону. Проводится специалистами исследования мочи, функции почек, лабораторная и рентгенологическая диагностика, анализ родословной. Лечение тубулопатии у детей чаще симптоматическое, направленное на нормализацию уровня калия, натрия, кальция и других микроэлементов. В данной статье вы сможете подробно ознакомиться с симптомами и особенностями терапии данного недуга.
Тубулопатии у детей
Не обращая внимания на то, что тубулопатиями считают заболевания, при развитии которых имеются нарушения функции почечных канальцев, некоторая часть нозологий этой группы являются на самом деле энзимопатиями — в их основе лежат нарушения структуры так называемых белков-переносчиков.
На практике частота встречаемости тубулопатии у детей сильно варьируется. Например, в медицине синдром Барттера крайне редко диагностируется, а вот частота почечных глюкозурий достигает почти 6% среди новорожденных малышей (учитывая семейную форму).
Актуальность данной проблемы чрезвычайно высока в педиатрии. Во-первых, тубулопатия почек у детей часто протекает под маской других болезней, по причине чего диагностируются они уже на стадии хронической формы почечной недостаточности, а она практически не поддается лечению. Во-вторых, причины основного количества первичных тубулопатий остаются не до конца изученными, а это значит, возможна лишь поддерживающая терапия. Клинические рекомендации тубулопатии у детей, однако, не стоит игнорировать.
Тубулопатии со вторичным гиперальдостеронизмом, сопровождающиеся потерей солей (ТСПС), обусловлены четко определенными наследственными патологиями канальцев почки. В патогенезе ТСПС в основном участвуют два сегмента дистального отдела нефрона: восходящее колено петли Генле (ВКПГ) и дистальный извитой каналец (ДИК). Функции сегментов до и после плотного пятна весьма различны, и это оказывает существенное влияние на клиническую картину патологий петли Генле и ДИКа — синдромов, подобных Барттеру и Гительману. Дефекты водонепроницаемой части восходящего колена, главная функция которого — реабсорбция солей, приводят к значительным потерям электролитов и воды, аналогично эффектам петлевых диуретиков (фуросемид). Напротив, дефекты в ДИКе, с его незначительной способностью к реабсорбции соли, учитывая его главную функцию (контроль экскреции кальция и магния), вызывают хронический дисбаланс электролитов, аналогичный последствиям хронического лечения тиазидами.
Наиболее тяжелое состояние развивается при сочетании патологий петли Генле и ДИКа. Оно сходно с усиленным мочегонным эффектом от совместного лечения петлевыми диуретиками и тиазидами. Помимо использования солевых растворов в терапии пациентов с данной патологией, ингибиторы простагландин E2-синтазы (например, чистый фуросемид) являются наиболее эффективным терапевтическим вариантом при полиурических расстройствах петли Генле, особенно у недоношенных детей с обезвоживанием и снижением ОЦК. При патологиях ДИКа блокаторы ренин-ангиотензин-альдостероновой системы (РААС), которые могут рекомендоваться после приема соли, калия и магния, считаются недостаточными. Похоже, что большинству пациентов с ТСПС на протяжении всей жизни требуется комбинация различных препаратов.
Наследственные тубулопатии: синдром Барттера и Гительмана
Синдромы Барттера и Гительмана являются редкими наследственными тубулопатиями, сопровождающимися потерей солей (ТСПС), что приводит к гипокалиемии. Они характеризуются дисфункцией трансэпителиального транспорта электролитов в восходящем колене петли Генле или дистальном извитом канальце (патологии ДИКа). Также встречается сочетание этих патологических процессов (комбинированные нарушения). Гипокалиемический алкалоз служит отличительной чертой патологий петли Генле и ДИКа и позволяет отличить их от тубулопатий, сопровождающихся потерей соли, в других отделах нефрона.
Синдром Гительмана является гораздо более распространенным заболеванием, чем синдром Барттера. В докладе из исследования Framingham Heart Study распространенность для синдрома Гительмана составляла 1:40 000 случаев по сравнению с 1:1 000 000 случаев для синдрома Барттера. Более низкая распространенность синдрома Барттера в популяции может быть частично обусловлена пренатальной или неонатальной смертностью, вызванной данной патологией, вследствие которой синдром не был диагностирован.
Общий обзор
Функции ВКПГ и ДИКа различны, а значит, патология этих сегментов нефрона вызывает совершенно разные изменения. Для оценки их отличий необходимо понимать основные механизмы транспорта электролитов и воды через эпителиальные клетки в трех основных частях дистальных канальцев: восходящее колено петли Генле, ДИК и дистальный отдел нефрона, чувствительный к альдостерону (ДОНЧА), включающий конечную часть ДИКа, трубочки, соединяющие ДИК и собирательные трубочки, и собирательные трубочки.
В восходящем колене петли Генле ключевым звеном в активном транспорте ионов Na+ и Cl– в клетки почечного канальца является чувствительный к фуросемиду натрий-калий-2-хлорид-котранспортер NKCC2. Ионы Na+ активно откачиваются из клеток восходящего колена петли Генле посредством базолатеральной Na+/K+-ATФазы, в то время как как Cl– покидает базальный полюс клетки через определенные каналы-переносчики ионов хлора (ClC-Ka и ClC-Kb). Для правильной работы обоих каналов-переносчиков ионов хлора требуется b-субъединица барттина. В отличие от ионов Na+ и Cl–, K+ повторно возвращается обратно в жидкость канальца через апикальную мембрану с помощью проницаемого для калия ионного канала — почечного наружного медуллярного калиевого канала (ПНМКК). Таким образом, неповрежденный ПНМКК необходим для создания положительного трансэпителиального потенциала просвета, который используется для активной реабсорбции трансцеллюлярной соли, а также для поддержания движущей силы пассивной парацеллюлярной транспортировки ионов Ca2+ и Mg2+ в восходящее колено петли Генле.
ДИК, который начинается после плотного пятна, играет важную роль в почечной экскреции хлорида натрия (около 10 % отфильтрованной нагрузки), а также кальция и магния (приблизительно по 8–10 % каждого). Как и в восходящем колене петли Генле, активный транцеллюлярный перенос соли в ДИКе обеспечивается за счет деятельности базолатеральной Na+/K+-АТФазы. В основе активного транспорта ионов Na+ в клетки в начале ДИКа лежит работа апикально экпрессированного тиазид-чувствительного натрий-хлор-котранспортера NCCT. В этой части ДИКа активность Na+/K+-АТФазы дополняется калиевым каналом внутреннего выпрямления Kir4.1, который облегчает базолатеральный выход ионов Na+. После внутриклеточного поглощения вместе с ионами Na+ ионы Cl– покидают клетку через ClC-Kb (ClC-Ka не экспрессирован в ДИКе). Как следствие нарушения транспорта ионов Na+, при любой патологии ДИКа внутриклеточная концентрация Na+ может быть понижена, что должно повысить поглощение ионов Na+ и увеличить экскрецию ионов Са2+ через клеточную мембрану в почечный интерстиций. Независимо от патогенеза гипокальциурия является отличительной чертой патологии ДИКа.
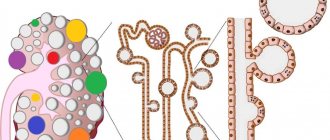
Рисунок 1 ❘ Реабсорбция растворенных веществ в восходящем колене петли Генле (ВКПГ), дистальном извитом канальце (ДИКе) и дистальном отделе нефрона, чувствительному к альдостерону (ДОНЧА), включая позднюю часть ДИКа, трубочки, соединяющие ДИК и собирательные трубочки, и собирательные трубочки.В ВКПГ(а) NaCl реабсорбируется фуросемид-чувствительным NKCC2 каналом вместе с калием, который затем рециркулирует через ROMK калиевые каналы. Кальций и магний пассивно реабсорбируются через парацеллюлярный путь. В ДИКе (b) реабсорбция соли происходит через котранспортер NCCT. Как и в ВКПГ, Na+ реабсорбируется в базолатеральном полюсе клетки с помощью Na+/K+-АТФазы, и Cl– покидает клетку через хлорные каналы. В этой части нефрона работа Na+/K+-АТФазы дополняется калиевыми каналами Kir4.1, которые облегчают базолатеральную реабсорбцию Na+ путем рециркуляции калия обратно в почечный интерстиций. Реабсорбция магния и кальция в ДИК является активной и обусловлена транспортом через селективные ионные каналы (TRPM6 и TRPV5 соответственно). В ДОНЧА калиевые каналы ROMK, в дополнение к их роли в ВКПГ, необходимы для секреции ионов калия в обмен на реабсорбцию натрия через эпителиальные натриевые каналы (ENaC) под влиянием альдостерона.
Классификация
Согласно патофизиологическиму и фармакологическому подходам, ТСПС могут быть разделены на две основные группы патологий канальцев почек: дефекты водонепроницаемой части восходящего колена, аналогичные эффекту петлевых диуретиков (фуросемид), и дефекты в ДИКе, аналогичные последствиям хронического лечения тиазидами. Первая группа далее подразделяется на патологии ВКПГ, вызванные генетическим дефектом в гене NKCC2 или гене ROMK (L1 и L2 соответственно). Вторая группа включает в себя нарушения, связанные с генетической патологией в каналах NCCT, ClC-Kb или Kir 4.1 (DC1, DC2 или DC3 соответственно). Генетические патологии ВКПГ также называются синдромом Барттера типа I и типа II, а патологии ДИКа — синдромом Гительмана, синдромом Барттера тип III и синдромом EAST. Тип L1 представляет собой фуросемид-подобную ТСПС, тогда как тип L2 представляет собой, прежде всего, фуросемид-подобную ТСПС с транзиторной гиперкалиемией из-за недостатка экспрессии ROMK канала в ДИКе, трубочках, соединяющих ДИК и собирательные трубочки, и собирательных трубочках, где это необходимо для секреции калия.
Комбинация патологий ДИКа и ВКПГ является одной из самых тяжелых категорий ТСПС, и это состояние образует третью группу: фуросемид-тиазидоподобную ТСПС (тип L-DC). Генетически это комбинированное расстройство относится к синдрому Барттера с сенсоневральной глухотой. В настоящее время эта группа патологий подразделяется далее на L-DC1 (синдром, вызванный комбинированными генетическими дефектами в каналах ClC-Ka и ClC-Kb) и L-DC2 (синдром, вызванный генетическим дефектом в b-субъединице барттина).
Рисунок 2 ❘ Сегмент в круглых скобках имеет второстепенное значение для клинического проявления. ADH — аутосомно-доминантная гипокальциемия; ASDN — дистальный отдел нефрона, чувствительный к альдостерону; BSDN — синдром Барттера с сенсоневральной глухотой; CaSR — кальциевый рецептор; DCT — ДИК; EAST/SeSAME — синдром, который сопровождает эпилепсия, атаксия, сенсорная глухота и тубулопатия; MAGE-D2 — меланома-ассоциированный антиген D2; taBS — переходный пренатальный синдром Барттера; TAL — ВКПГ.
Генетика и клинические проявления
Различные варианты синдрома Барттера — редкие наследственные нарушения, которые охватывают гетерогенную группу ТСПС, поражающих ВКПГ, с разнообразными причинами, но с общей точкой патогенеза — потерей функции NKCC2 канала. Среди фенотипов синдрома Барттера наиболее распространены мутации, которые влияют на ген KCNJ1 (канал ROMK), реже — на ген SLC12A1 (канал NKCC2), ген CLCNKB (канал ClC-Kb) и ген BSND (белок барттин). Нефрокальциноз типичен для фенотипов с мутациями генов KCNJ1 и SLC12A1. Хроническая почечная недостаточность, хоть встречается и редко, но все же обнаруживается среди фенотипов с мутациями в генах KCNJ1, CLCNKB и BSND (при этом нефрокальциноз встречается не всегда). Потеря слуха происходит только у пациентов с мутациями гена BSND.
Синдром Гительмана, также известный как семейная гипокалиемия с гипомагниемией, является одной из наиболее частых наследственных тубулопатий и характеризуется гипокалиемическим метаболическим алкалозом со значительной гипомагниемией и гипокальциурией. Болезнь вызвана мутациями гена SLC12A3, расположенного в локусе 16q13 16-й хромосомы, и имеет аутосомно-рецессивный тип наследования. Ген SLC12A3 кодирует NCC белок, экспрессирующийся в начальной части ДИКа, функция которого подавляется тиазидными диуретиками.
Рисунок 3 ❘ При всех перечисленных заболеваниях присутствует гипокалиемия и алкалоз. Стрелки вверх и вниз указывают соответственно на увеличение или уменьшение экскреции или концентрации. Знаки в круглых скобках указывают на то, что либо данный патофизиологический признак является вторичным и протекает в легкой форме, и/или изменения редко присутствуют, и/или незначительно отличаются от нормальных показателей. n. d. — не определено. CaSR — кальциевый рецептор; ClC — хлорный канал; HAS — гиперальдостеронизм; HPU — гиперпростагландинурия; MAGE-D2 — меланома-ассоциированный антиген D2; NCC — хлорангидрид натрия; NDI — нефрогенный несахарный диабет; NHPT — гиперпаратиреоз новорожденных; NKCC2 — натрий-калий-2-хлорид-котранспортер; PH — полигидрамнион; РОМК — внешний медуллярный калиевый канал.
Терапия
Общим в терапии как патологий ВКПГ, так и патологий ДИКа является восполнение потерянной жидкости (насколько это возможно). Цель должна состоять в том, чтобы восстановить баланс электролитов, пул минеральных веществ в организме и объем внеклеточной жидкости. Это самый эффективный способ лечения метаболического алкалоза. В остальном подход к лечению патологий ДИКа и ВКПГ значительно отличается. У младенцев с жизнеугрожающими патологиями ВКПГ восстановление баланса воды и электролитов (предпочтительно через центральный венозный катетер) является первой терапевтической мерой для предотвращения шока и острой почечной недостаточности. Однако необходимо понимать, что чрезмерная гемодилюция усугубит полиурию. Поскольку канальцевый ток мочи связан, по крайней мере частично, со стимуляцией синтеза почечного PGE2, показана антидиуретическая терапия с ингибитором синтеза PGE2 (например, индометацин). Из-за растущей уязвимости слизистой оболочки желудка к неселективным нестероидным противовоспалительным средствам (НПВС) селективные ингибиторы циклооксигеназы-2, например, целекоксиб, можно рассматривать в качестве терапии для детей и молодых взрослых с патологиями желудочно-кишечного тракта. У детей старшего возраста и взрослых пациентов с медленно развивающейся патологией ДИКа диета, богатая солью и калием, является основой краткосрочного и долгосрочного лечения. Однако следует отметить, что терапия с использованием магния предпочтительна в виде модифицированных органических солевых препаратов; она рекомендуется для эффективного лечения и профилактики осложняющей основное заболевание гипокалиемии. В частности, у пациентов с патологиями ДИКа и хондрокальцинозом, очевидно, связанным с гипомагниемией, отмечены улучшения в состоянии при назначении магния. Кроме того, после назначения магния значительно улучшается состояние беременных женщин, больных синдромом Гительмана, особенно с гиперемезисом (неукротимой рвотой).
Источники
1. Seyberth H. W., Weber S., Kömhoff M. Bartter’s and Gitelman’s syndrome //Current opinion in pediatrics. – 2020. – Т. 29. – №. 2. – С. 179-186. 2. Naesens M. et al. Bartter’s and Gitelman’s syndromes: from gene to clinic //Nephron Physiology. – 2004. – Т. 96. – №. 3. – С. p65-p78. 3. Koulouridis E., Koulouridis I. Molecular pathophysiology of Bartter’s and Gitelman’s syndromes //World Journal of Pediatrics. – 2020. – Т. 11. – №. 2. – С. 113-125.
Причины тубулопатий
Практически все тубулопатии возникают по причине генетического дефекта. Мутации могут происходить в генах, которые кодируют синтез белков, которые транспортируют разные микроэлементы сквозь мембрану клеток канальцев почек.
Также имеет место быть отсутствие реакций на гормоны, которые регулируют реабсорбцию натрия, калия, бикарбонатов, а также других соединений, а это связано с дефектами развития рецепторов к данным гормонам на клеточной мембране.
В протоколе тубулопатии у детей сказано, что еще возможной причиной считаются диспластическое изменение в почках — неправильная закладка ткани во внутриутробное время, по причине чего изменяются сами структуры мембран тубулоцитов.
Все вышеперечисленные факторы способны привести к нарушениям процесса реабсорбции разных соединений, а также микроэлементов из мочи. Но то же самое способно происходить и в почечном канальце после отравлений солью тяжелых металлов, лекарственными средствами, при болезнях накопления (например, мукополисахаридоз, сфинголипидозы и др.), когда имеет место также генетический дефект, который находится за пределами почек, а также мочевыделительной системы.
Любое заболевание, способное привести к нарушениям функционирования почечного канальца, может спровоцировать тубулопатии. Сюда также следует отнести ожог тяжелой степени, заболевания крови, а также другие состояния, во время которых значительно повышается нагрузка на почки.
Осложнения и профилактика
Основное и наиболее распространенное негативное последствие патологии – развитие острой недостаточности органа. Более редко диагностируют некроз коркового вещества почек. Причины таких осложнений – нарушение процессов обмена и формирование почечной ишемии.
Особую осторожность необходимо соблюдать тем лицам, у которых имеется повышенный риск развития патология в силу наследственной предрасположенности. Таким людям рекомендовано систематически посещать врача для прохождения медицинских обследований, чтобы своевременно выявить болезни почек и начать соответствующую терапию.
Классификация тубулопатий
Разделение на первичную и вторичную тубулопатию у детей основано на этиологических принципах. В первой ситуации идет речь о наследственной и врожденной патологии в системе канальцев почек, а вторичные тубулопатии связаны напрямую с воздействием извне: с заболеваниями другого органа и системы, болезнями крови, ожоговой болезнью, опухолями и т.д.
Помимо этого, существует и клиническая классификация, которая основана на ведущем синдроме недуга: скелетных деформациях, полиурии, нефролитиазе и других признаках. На практике используется часто классификация по уровню поражения канальцевого аппарата. Выделяются следующие тубулопатии:
- с сильным поражением проксимального канальца (цистинурия, почечная глюкозурия, болезнь де Тони-Дебре-Фанкони и др.).
- с сильным поражением дистального канальца (псевдогипоальдостеронизм, почечный тубулярный ацидоз первого типа).
- с нарушениями реабсорбции на уровне собирательной трубочки (синдром Лиддла, гиперальдостеронизм, синдром Барттера).
- с тотальным повреждением всего канальцевого аппарата (нефронофтиз).
Какие причины способствуют развитию заболевания
Причины возникновения тубулопатии будут отличаться, исходя из ее формы. Так, различают первичную форму заболевания и вторичную. Первичная тубулопатия развивается, если:
- Мембраны, которые переносят белковые вещества, имеют нарушенную структуру.
- Ферменты, доставляющие питательные вещества в мочевыделительные органы, имеются в недостаточном объеме.
- Канальца и мембраны имеют низкую чувствительность к действию гормонов.
Вторичная тубулопатия развивается вследствие повреждения транспортных канальцев на фоне генетики. Иными словами, если женщина имеет такую патологию в организме, шанс ее развития у будущего ребенка значительно повышается. Иными причинами возникновения вторичной формы заболевания можно назвать:
- Развитие любого воспалительного процесса, который ввел в поражение почечную ткань.
- Развитие дисплазии органа.
- Наличие в организме обменных патологий.
Заболеванию подвержены люди любого возраста, включая новорожденных и пожилых. Если предотвратить врожденную тубулопатию невозможно, то в случае с приобретенной формой заболевания можно направить все силы на снижение риска его возникновения в будущем.
Симптоматика
Итак, мы с вами разобрали, что чаще всего на практике встречаются наследственные тубулопатии у детей. Но каковы же их симптомы?
Клинические проявления будут зависеть от того, транспорт какого конкретно соединения нарушается. Например, при гипофосфатемическом диабете будет наблюдаться избыточное выведение кальция с мочой, по причине чего в клинике превалирует симптоматика рахита, высокорезистентного к лечению витамином D.
Также следует отметить, что рахитоподобное изменение развивается и при заболевании де Тони-Дебре-Фанкони, однако в данном случае оно сочетается с костными множественными деформациями, а также патологиями со стороны какого-либо другого органа (сердца и сосудов, глаз, ЛОР-органов и других).
Псевдогипоальдостеронизм определяется по избыточному выведению хлорида натрия из организма больного, причем и с мочой, и с калом, а также слюной, поэтому у пациентов наблюдаются признаки обезвоживания.
Дефицит в крови глюкозы при тубулопатии проявляется в виде слабости, чувства голода, полиурии, полидипсии, по причине чего глюкозурия почечная сопровождается признаками, напоминающими заболевание сахарным диабетом.
Сокращение реабсорбции калия с гипокалиемией может быть причиной мышечной гипотонии, а также развития параличей. Нарушения мембранного почечного транспорта аминокислот провоцируют их дефицит, который проявляется теми или иными признаками. Например, при цистинурии появляются почечные колики, при заболевании Хартнапа – симптомы мозжечковой атаксии, а также пеллагры. Эта группа тубулопатий приводит часто к нефролитиазу.
Как видите, симптомы тубулопатии у детей могут быть абсолютно разными.
Ведущий синдром
В зависимости от ситуации могут проявляться следующие синдромы и поражения.
Наличие полиурии
Глюкозурия – одно из проявлений тубулопатии
В данном случае первичная тубулопатия проявляется в несахарном диабете почечного характера и почечной глюкозурии.
Также наблюдается увеличение количества глюкозы, как в моче, так и в крови. Симптоматика практически не вызывает беспокойства у больного, поэтому выявить болезнь на ранних этапах бывает сложно.
В данном случае специалисты рекомендуют пить больше жидкости, для того, чтобы восполнить ее потерю в организме.
Вторичная тубулопатия проявляется в протекании:
- нефронофтиза Фанкони;
- пиелонефрита;
- цистиноза;
- тирозинемии;
- хронической почечной недостаточности.
Лечение этих недугов проводиться медикаментозными препаратами. Чтобы поддержать самочувствие больного необходимо регулярно наблюдаться у врача-нефролога и сдавать анализы крови и мочи.
Развитие аномалий в человеческом теле
Аномалии в развитии могут проявиться совместно со следующими заболеваниями:
- Первичные тубулопатии сопровождаются протеканием фосфат-диабета, синдрома Тони-Дебре-Фанкони и почечного тубулярного ацидоза. Лечение в данном случае направленно на устранение первоначальных причин появления заболевания. Если удается справиться с сопутствующими недугами, лечение нарушения происходит проще.
- В случае вторичного нарушения может развиваться витамин-Д зависимый рахит, целиакия и гипофосфатазия. При протекании таких болезней целесообразно использовать для лечения витамин Д. Для получения положительного эффекта дозы препараты должны быть максимально высокими. При этом процесс лечения ведется под строгим контролем специалиста. Больному так же необходимо придерживаться правильного питания с ограничением соленой пищи.
Развитие нефролитиаза
В данном случае тубулопатия сопровождается такими симптомами и синдромами:
- При первичном заболевании диагностируется цистинурия, глицинурия и иминоглицинурия. Заболевание легко держать под контролем. Обязательным условием лечения является соблюдение рекомендаций врача и регулярная сдача лабораторных анализов мочи и крови. При наличии отклонений, лечение может проводиться в условиях стационара.
- Вторичная разновидность нарушения сопровождается протеканием оксалоза, ксантинурии и синдрома Леша-Нихана. Для нормальной работы почек применяется лечение медикаментозными препаратами, которые в свою очередь направлены на уменьшение проявления признаков сопутствующих заболеваний.
Диагностика
При подозрении на тубулопатии педиатры назначают комплексное обследование мочи и функции мочевыделительной системы. Также обязательным будет определение уровня кальция, натрия, калия, хлора в моче, тест на глюкозу.
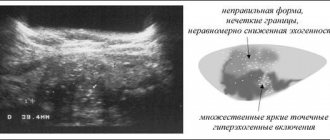
Обнаружить в анализе аминокислоты можно при помощи хроматографии. Конкременты будут исследоваться визуально с помощью микроскопа или выявляются при рентгенографическом исследовании и УЗИ.
УЗИ-диагностика подтверждает часто аномалии почек, мочеточников, а также других отделов в мочевыделительной системе. Для большинства первичных тубулопатий устанавливается тип наследования, локализация дефектов в хромосоме, по причине чего необходим анализ всей родословной.
Содержание разных микроэлементов устанавливается в моче, в крови. При повышенной экскреции отмечается их дефицит. Биохимические анализы крови позволяют оценить показатель глюкозы. При тубулопатии определяется также количественное содержание в крови гормонов. Внимание уделяется гормону щитовидной железы и гормону надпочечников.
Лечение тубулопатии
Так как патология обусловлена чаще всего генетическими дефектами, не разработана до сих пор этиотропная терапия. Исключение составляют только вторичные тубулопатии.
В таких случаях коррекция предрасполагающих факторов приводит к улучшению состояния. При ожогах тяжелой степени проводят гемодиализ, при лизосомных заболеваниях накопления показаны заместительные ферментные методы терапии, предупреждающие отложение разных соединений в почечном канальце. Независимо от главной причины тубулопатии врачом назначается так называемая ощелачивающая диета, включающая молочные продукты, картофель, капусту и фрукты.
Общая информация
Тубулопатии – группа определенных заболеваний почек, имеющих схожий механизм развития, не отличаются клиническими проявлениями и методом терапии.
В медицине различают несколько типов тубулопатий в зависимости от причины их появления и периода возникновения.
Тубулопатии возникают в основном по причинам нарушения формирования плода или под влиянием различных факторов, оказывающих влияние на структуру почек.
Симптомы неспецифичны и для установления точного диагноза требуется проведение тщательной диагностики.
Терапия первичных тубулопатий
Терапия первичных тубулопатий направлена на нормализацию показателя микроэлементов, которые выводятся усиленно с мочой. Дефицит корригируется введением внутривенно раствора хлорида натрия, препаратов калия, а также цитратных смесей.
В терапии рахита применяются высокое количество витамина D, бисфосфонаты, которые способствуют задержке кальция, фосфора в организме больного. Выраженная костная деформация является показанием к хирургическому вмешательству. Так как при тубулопатиях отмечается часто задержка физического развития, имеет место назначение курса синтетического соматотропина.
Итог болезни
Прогнозы не всегда положительные, зависят от этиологии возникновения недуга. Формирование несахарного почечного диабета у детей приводит к быстрому летальному исходу. Наблюдается обезвоживание, которое является причиной смерти. При синдроме де Тони-Дебре-Фанкони появляется почечная недостаточность, которая требует заместительной терапии.
Но не все так пессимистично. Большинство тубулопатий поддается лечению. Однако некоторые заболевания протекают бессимптомно. Именно поэтому важно регулярно проводить обследование ребенка.
Клиническая картина
Клинические проявления заболеваний многообразны и зависят от того, какой элемент мочевыделительной системы был затронут.
В некоторых случаях наблюдается выведение большого количества кальция вместе с уриной. Рахитоподобные нарушения сочетаются с деформацией глаз, сосудов, сердца, ушей, носа. Также отмечаются признаки обезвоживания.
При недостаточном количестве глюкозы возникают следующие симптомы:
- чувство голода;
- слабость;
- онемение конечностей.
Данные признаки часто напоминают сахарный диабет, в результате чего многие пациенты ищут причину их появления не в той области.